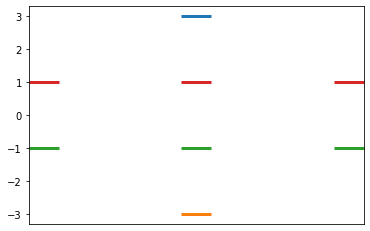
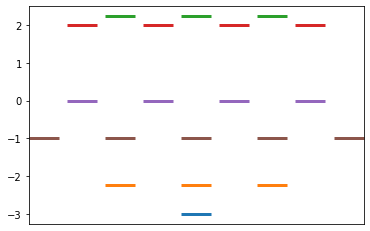
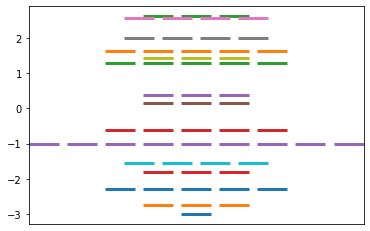
Constructs the Huckel matrix and solves for the values, based on Ex 3.1 instructions
This solver will only work if all \(p\) orbitals are equivalent (it will not consider for instance perturbations due to nitrogen in pyridine). The Huckel theory assumptions are: \[ H_{ij}= \begin{cases} \alpha,\ i=j\\ \beta,\ i \text{ adjacent to } j\\ 0,\ \text{otherwise}\end{cases} \] with scaling such that \(\alpha = 0\) and \(\beta =-1\). As such, it can be seen that the resulting Huckel matrix is the negative of the adjacency matrix \(A\) where: \[ A_{ij}= \begin{cases} 1,\ i \text{ adjacent to } j\\ 0,\ \text{otherwise}\end{cases} \]
generate the smiles of either a straight chain or ring polyene, with n atoms. All carbons will be sp2 hybridised. For linear molecules with an odd number of atoms it will return the anion For rings with 4n+1 atoms it will return the anion eg C5H5- For rings with 4n+3 atoms it will return the cation eg C7H7+
From a Huckel matrix input, solve for the Huckel pi system Returns a dictionary of energy levels with the associated (possibly degenerate) wavefunctions
# rdkit does consider all bonds, even if all are not shown on the output skeletal diagram: this is mostly apparent for cubanemolecule = Chem.MolFromSmiles(cubane_smiles)mat =-Chem.GetAdjacencyMatrix(molecule)print(mat)